VCFfiles¶
20200114 VCF2PopTree¶
Reference: PeerJ
Github download: VCF2PopTree
MEGA1
Minimum quality score: 80
Minimum coverage depth: 100
Distance: Genetic Distance
Missing Data: Use SNVs for each pair of genomes
Model: Number of differences
Construct Tree: UPGMA Tree
Output format: Pair-wise diversity(MEGA)
Select genomes: All
****
Basics in VCF Notes¶
Aims for this notes¶
- Explanation for my own VCF files in different version;
- Understand better about my data before moving to downstream analyses.
Basics in VCF¶
VCF: Variant Call Format It is a standardized text file format for representing SNP, indel, and structural variation calls.
SNP: Single nucleotide polymorphisms Definition

Assembly: here should be a link added later to study with assembly
- De novo assembly from the start will get a integrated genome from one chromosome;
- SNPs happens after mapping(alignment) step – and is two chromosomes (diploid) or more (polyploid) with two strands of DNA. Those showed in SNP data files as ‘heterozygosity’ means there’re two or more different types of nucleotides in one site, it might be: 1) sequence error; 2) difference from maternal and paternal (the true consequence of ‘heterozygosity’); 3) SNP from DNA duplication.
okay, still confused here, look back later. Main struggling here is DNA strands, chromatids, chromosomes, etc. Might get answers after learning the assembly stuff, now continue the VCF file first
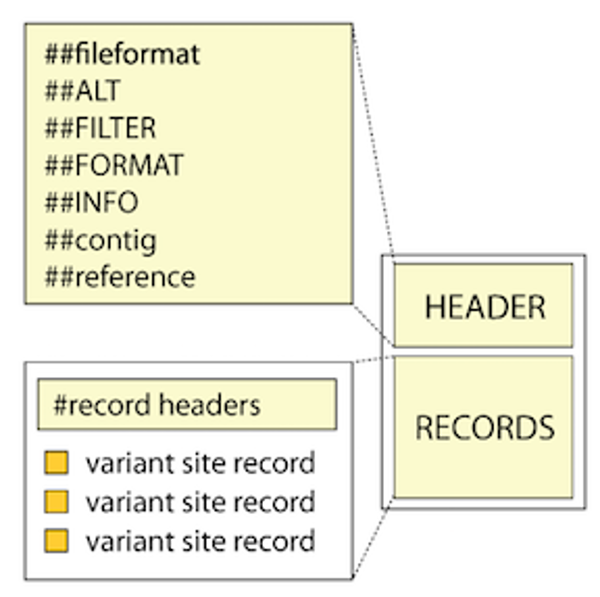
VCF file from TASSEL-GBS¶
Header¶
##fileformat=VCFv4.0
##Tassel=<ID=GenotypeTable,Version=5,Description="Reference allele is not known. The major allele was used as reference allele">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=AD,Number=.,Type=Integer,Description="Allelic depths for the reference and alternate alleles in the order listed">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth (only filtered reads used for calling)">
##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Genotype Quality">
##FORMAT=<ID=PL,Number=.,Type=Float,Description="Normalized, Phred-scaled likelihoods for AA,AB,BB genotypes where A=ref and B=alt; not applicable if site is not biallelic">
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=AF,Number=.,Type=Float,Description="Allele Frequency">
##fileformat is the VCF version, sometimes will influence the downstream analyses;
##TASSEL is similar as ##FILTER in elsewhere, I’m not quite sure why here is no information of reference allele.
##FORMAT & ##INFO define the annotations contained in the FORMAT & INFO columns of the VCF file, details are in the following.
Variant call records¶
For each site record, the information is structured into columns (also called fields) as follows:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT E4E0324_2 [other samples...]
The first 8 columns represent the properties observed at the level of the variant site. Sample-specific information such as genotype and individual sample-level annotation values are contained in the FORMAT column (9th column) and in the sample-name columns (10th beyond).
Site-level properties and annotations
These first 7 fields are required by the VCF format and must be present, although they can be empty (in practice, there has to be a dot, ie . to serve as a placeholder)
- CHROM and POS: The contig and genomic coordinates on which the variant occurs.
- ID: An optional identifier for the variant.
- REF and ALT: The reference allele and alternative allele(s) observed in a sample, set of samples, or a population in general (depending how the VCF was generated). Note that REF and ALT are always given on the forward strand. so, how to know how the VCF generated to get this information??
- QUAL: The Phred-scaled probability that a REF/ALT polymorphism exists at this site given sequencing data. The larger, the more possibility of variant it could be – but not very reliable.
- FILTER: This field contains the name(s) of any filter(s) that the variant fails to pass, or the value
PASSif the variant passed all filters. If the FILTER value is., then no filtering has been applied to the records.
This next field does not have to be present in the VCF.
- INFO: Various site-level annotations.
Sample-level annotations
The short names of the sample-level annotations are recorded in the FORMAT field.
- GT-the Genotype of this sample at this site:
0means the same asREF,1means the firstALT,2means the secondALT, (and so on). For diploid:
0/0means the sample is homozygous reference0/1means the sample is heterozygous, carrying 1 copy of each of the REF and ALT alleles1/1means the sample is homozygous alternate./.means missing data
AD and DP: Allele depth and depth of coverage: These are complementary fields that represent two important ways of thinking about the depth of the data for this sample at this site.
AD is the unfiltered allele depth, i.e. the number of reads that support each of the reported alleles. In diploids, two values seperated by
,, the former is REF, the latter is ALT.DP is the filtered depth, at the sample level. This gives you the number of filtered reads that support each of the reported alleles.
GQ-Quality of the assigned genotype: represents the Phred-scaled confidence that the genotype assignment (GT) is correct, derived from the genotype PLs. The likelihood of second most likely variants, maximum is 99. The larger, the more possibility of supporting the best one is the ‘best’.
PL-“Normalized” Phred-scaled likelihoods of the possible genotypes: for monomorphic site (which only has one ALT) in diploids, three numbers are given for evaluate the possibility of 0/0, 0/1, 1/1; “normalized” is quotes here because it’s not real ‘probabilities’, just a way to show possibilities. For this, 0 is the most likely ones (which also showed in GT, if it’s right)
examples in my VCF
First, no variant:
- E4E0072_3 the first site:
GT:AD:DP:GQ:PL 0/0:5,0:5:96:0,15,180At this site, the called genotype is
GT=0/0, which corresponds to a homozygous reference genotype with allelesT/T;The confidence indicated by
GQ=96is good; means the second likely genotype is very low possibility, GT is reliable enough, also the degree of certainty in our genotype is evident in the PL field, wherePL(0/0)=0(the normalized value that corresponds to a likelihood of 1.0) as is always the case for the assigned allele, the next PL isPL(0/1)=15, corresponding to 10^(-1.5), or 0.0316, actually is not very good, means there’re still has the possibility to beT/C; but the next one is very small, means there’re nearly impossible to beC/C.Others, there were a total of 5 informative reads at this site (
DP=5), all of which supported the REF allele (=had the reference base) (indicated byAD=5,0).So, the conclusion is the genotype in this sample at this site is homozygous reference, no variant but has the possibility that it would be heterozygous, which is
T/Cit might because the reads coverage is too low (only 5), but it’s impossible to beC/C.
Then, this variant is weird:
- AT1627_2 at the the site
16168at chromosomeNODE_1007_LENGTH_33384_COV_15.053492:GT:AD:DP:GQ:PL 2/2:0,0,2:2:33:0,0,0,255,255,255
GT=2/2means genotype isA/A, homozygous second alternate;
AD=0,0,2&DP=2means there were in total 2 informative reads at this site, all of which supported the second ALT allele (=had the alternate base); – with so little coverage, we can not be sure that the genotype should not in fact be homozygous second alternate variant.
GQ=33means the confidence is not very good, corresponding to 10^(-3.3), or 0.0005, indicating that even though this is probably a real variant (the QUAL is good), we are not sure we have the right genotype.Here comes the most weird thing:
PL(0/0:0/1:1/1)=0:0:0whilePL(0/2:1/2:2/2)=255:255:255==>> HOW CANGTBE2/2?