Tetraploid reference¶
Here, we compare different samples (DIP/TET/ALL) with different scaffolds (conserved/tetraploid/all) of E. arctica as reference genome
TASSEL-GBS (SNP Calling)¶
Essential files¶
FASTAQ: /disk2/Twyford_GBS_illumina
KEYFILE:
Tetraploids: /localdisk/home/s1950737/SNPCall/keyfile/TetKey.txt
Diploids: /localdisk/home/s1950737/SNPCall/keyfile/DipKey.txt
ALL: /localdisk/home/s1950737/SNPCall/keyfile/AllKey.txt
REFERENCE GENOME: /disk2/hbecher_tmp/remap/scaffolds_disom_concat_dipOrTet.fasta
SCAFFOLDS FILTER:
Conserved scaffolds: /loaldisk/home/s1950737/SNPCall/scaffolds/uni.tsv
Tetraploid scaffolds: /loaldisk/home/s1950737/SNPCall/scaffolds/tet.tsv
Steps¶
Get SAM files:TASSEL-GBS PIPELINE
Step1: GBSSeqToTagDBPlugin
./run_pipeline.pl -Xms100G -Xmx200G -GBSSeqToTagDBPlugin -db /FILEPATH/GBS.db -e ApeKI -i /disk2/Twyford_GBS_illumina -k /FILEPATH/KEYFILE.txt
Step2: TagExportToFastqPlugin
./run_pipeline.pl -TagExportToFastqPlugin -db /FILEPATH/GBS.db -o /FILEPATH/tagsForAlign.fa.gz -c 1 -endPlugin
Step3: Alingment
Step3-1: create an index from the reference genome
cp /disk2/hbecher_tmp/remap/scaffolds_disom_concat_dipOrTet.fasta .
./bwa index -a bwtsw ../referenceGenome/tet/scaffolds_disom_concat_dipOrTet.fasta
Time consumed: 16:30 13/04/20 - 17:00 13/04/20; 30 minute
Step3-2: Aligment
./bwa aln -t 4 ../referenceGenome/tet/scaffolds_disom_concat_dipOrTet.fasta ../FILEPATH/tagsForAlign.fa.gz > tagsForAlign.sai
./bwa samse ../referenceGenome/tet/scaffolds_disom_concat_dipOrTet.fasta tagsForAlign.sai ../FILEPATH/tagsForAlign.fa.gz > tagsForAlign.sam
Filter SAM files with SCAFFOLDS FILTER TXT-samtools
samtools view -bh tagsForAlign.sam > tagsForAlign.bam
samtools sort tagsForAlign.bam > OUT.bam
samtools index -c OUT.bam
cat uni.tsv | tr "\n" " " | xargs samtools view -h OUT.bam > output.sam
SNP calling
Step4: SAMToGBSdbPlugin
./run_pipeline.pl -SAMToGBSdbPlugin -i ../FILEPATH/output.sam -db ../FILEPATH/GBS.db
Step5: DiscoverySNPCallerPluginV2
./run_pipeline.pl -DiscoverySNPCallerPluginV2 -db ../FILEPATH/GBS.db
Step6: ProductionPipeline ProductionSNPCallerPluginV2
./run_pipeline.pl -Xms100G -Xmx200G -ProductionSNPCallerPluginV2 -db ../FILEPATH/GBS.db -e ApeKI -i /disk2/Twyford_GBS_illumina -k ../FILEPATH/KEYFILE.txt -o ../FILEPATH/OUTPUT.vcf
TIME CONSUMED¶
| DATA | SCAFFOLDS | STEP1 | STEP2 | STEP3-2 | STEP4 | STEP5 | STEP6 | TOTAL | DATE |
|---|---|---|---|---|---|---|---|---|---|
| TET | conserved | 70m | 12m | 6m | 1m | 20m | 38m | 2.45h | 14/04/20 |
| TET | tetraploid | 43m | 6m | 4m | 3m | 3h | 40m | 4.6h | 29/04/20 |
| DIP | conserved | 11m | 1m | <1m | 10s | 6m | 12m | 0.5h | 15/04/20 |
| ALL | conserved | 47m | 13m | 2m | 1m | 25m | 56m | 2.4h | 28/04/20 |
| ALL | all | 40m | 14m | 4m | 3m | 1d13h20m | 63m | 1.5d | 28/04/20 |
PLINK (SNP Filtering) & DAPC TEST¶
FILTERING¶
Check the missing data
make missing data with individuals & sites
Setting the rule: keep ~90% taxa & 20% sites, maf = 0.05, thus, for samples, ALL(776/862); TET(704/782); DIP(72/80)
| ITEM | vcf(size) | vcf.gz | bi-vcf | SITES | SITES-LEFT | INI-FILTER | SIZE | LESS-FILTER | SIZE |
|---|---|---|---|---|---|---|---|---|---|
| ATA | 5.84GB | 328.9MB | 6.01GB | ||||||
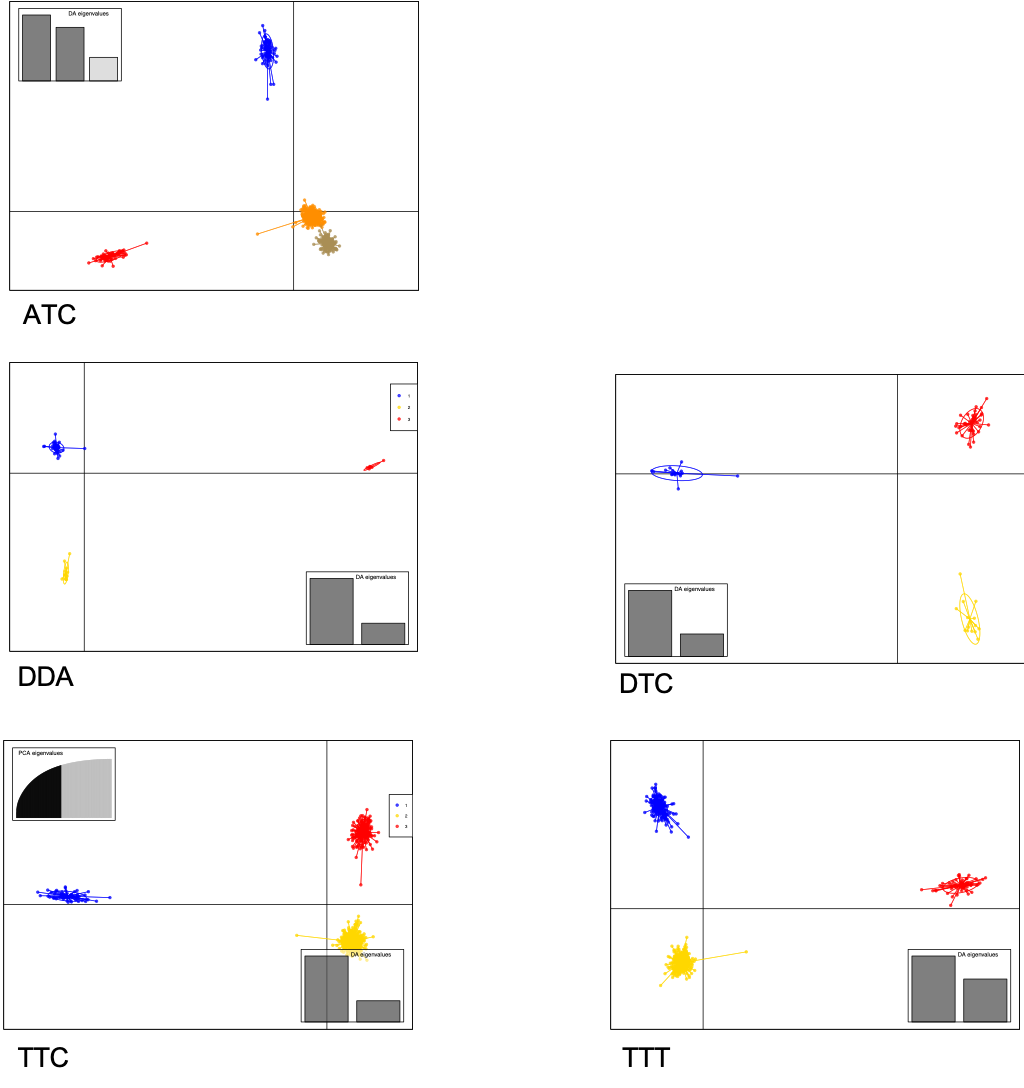
| ATC | 360.9MB | 19.6MB | 394.9MB | 27,513(26,623) | ~5502(5324) | 0.80/0.35/0.05 | 21MB | 0.75/0.20/0.05 | 11.5MB |
| DDA | 143.5MB | 9.9MB | — | 100,903 | ~20,180 | 0.72/0.25/0.05 | 10.9MB | 0.72/0.10/0.05 | 5.9MB |
| DTC | 14.4MB | — | — | 10,035 | ~2,007 | 0.75/0.25/0.05 | 1.2MB | 0.75/0.20/0.05 | 993KB |
| TTC | 346.6MB | 17.4MB | 335.9MB | ~(4989) | 0.80/0.35/0.05 | 18MB | 0.75/0.20/0.05 | 9.8MB | |
| TTT | 844.8MB | 46.5MB | 924.4MB | 70,844(68,574) | ~14,168(13,714) | 0.80/0.35/0.05 | 49.5MB | 0.75/0.20/0.05 | 27.3MB |
For those contain tetraploid samples (TET/ALL), should exclude more than 2 alleles sites first:
vcftools --vcf VCF.vcf --min-alleles 2 --max-alleles 2 --recode --out bi-VCF
Then, use plink to finish the initial filter/less missing data filter:
./plink --file ITEM.plk --allow-extra-chr --mind 0.75 --maf 0.01 --geno 0.10 --recode vcf-iid --out ITEM-INI
Then, using less-missing data to do the DAPC(R):
library(vcfR)
library(poppr)
library(ape)
library(RColorBrewer)
library(reshape2)
library(ggplot2)
# load and check out vcf data set:
lm.vcf <- read.vcfR('./LessMissing/ATC-LESS.vcf')
# turn into the vcf data to genlight:
gl.lm <- vcfR2genlight(lm.vcf)
ploidy(gl.lm) <- 2
#this is interactively choose the K, use Kmeans as default
grp_lm_k <- find.clusters(gl.lm, n.pca = 300) #interactive, pca = 300; k=3
plot(grp_lm_k$Kstat, type="b", xlab="number of clusters (K)", ylab="BIC", col="blue", main="Detection based on BIC")
points(3, grp_lm_k$Kstat[3], pch="x", cex=2)
mtext(3, tex="'X' indicates the actual number of clusters")
# check out in find.cluster groups
x1 <- grp_lm_k$grp #k=3
write.csv(x1, file = 'ATC-K3.csv')
## dapc
lm.dapc <- dapc(gl.lm, pop = factor(x1), n.pca = 30, n.da = 100) ##not quite sure about n.da
scatter(lm.dapc, cex = 1.5, legend = TRUE, clabel = F, scree.pca = FALSE, posi.da = "bottomright", cleg = 0.75)
TEST DIFFERENT PARAMETERS¶
KEY POINT: kmer=80, change step: step 1(GBSSeqToTagDBPlugin) & step 5(DiscoverySNPCallerPluginV2)
DETAILS:
- STEP1(GBSSeqToTagDBPlugin):
P1: 1. Min Kmer Count(-c=30);
P1: 2. Minimum quality score (-mnQS=30);
P1, P2: 3. Minimum Kmer length (-kmerlength=80)
- STEP2(TagExportToFastqPlugin):
- P1: 1. Min count (-c=5)
- STEP5(DiscoverySNPCallerPluginV2)
P1: 1. Max tags per cut site position (-maxTagsCutSite=80);
P1: 2. Minimum Minor Allele Freq (-mnMAF=0.05)
- Production
P1, P2: 1. length of Kmer (-kmerLength=80);
P1: 2. minimum quality score (-mnQS=30)
TEST DATASET: DIP-TET-CON
| DATA | STEP1 | STEP2 | STEP3-2 | STEP4 | STEP5 | STEP6 | TOTAL | DATE | SIZE | SITES | LEFT | INITIAL-FILTER |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
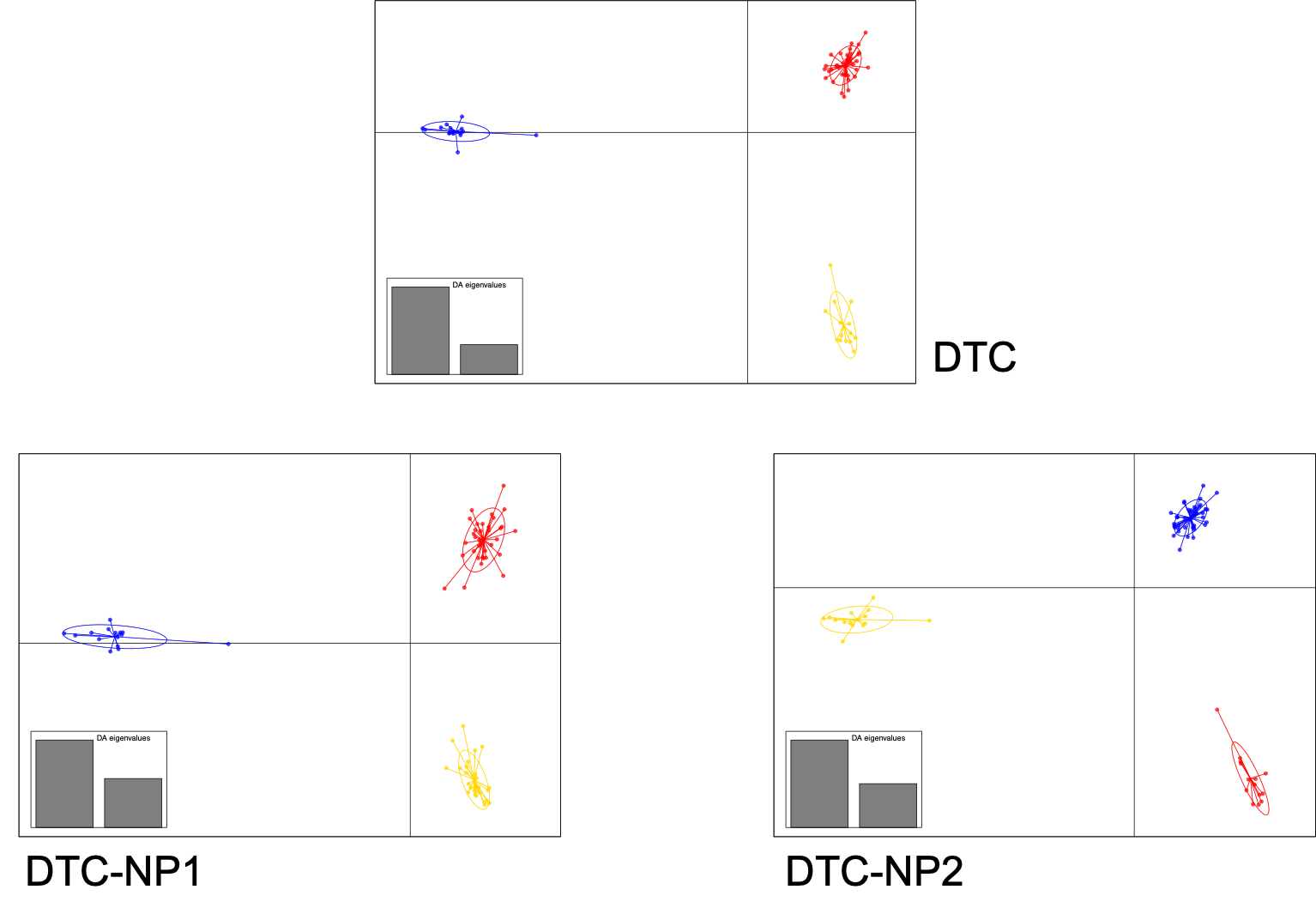
| DTC | 11m | 1m | <1m | 10s | 6m | 12m | 0.5h | 14/04/20 | 14.4MB | 10,035 | 2,007 | 0.75/0.25/0.05 |
| DTC-P1 | 17m | <1m | 17s | 5s | 4m | 15m | 0.55h | 06/05/20 | 5.7MB | 3,714 | 1,500 | 0.78/0.30/0.05 |
| DTC-P2 | 17m | <1m | <1m | 5s | 6m | 17m | 0.6h | 06/05/20 | 16.2MB | 11,297 | 2,260 | 0.75/0.25/0.05 |
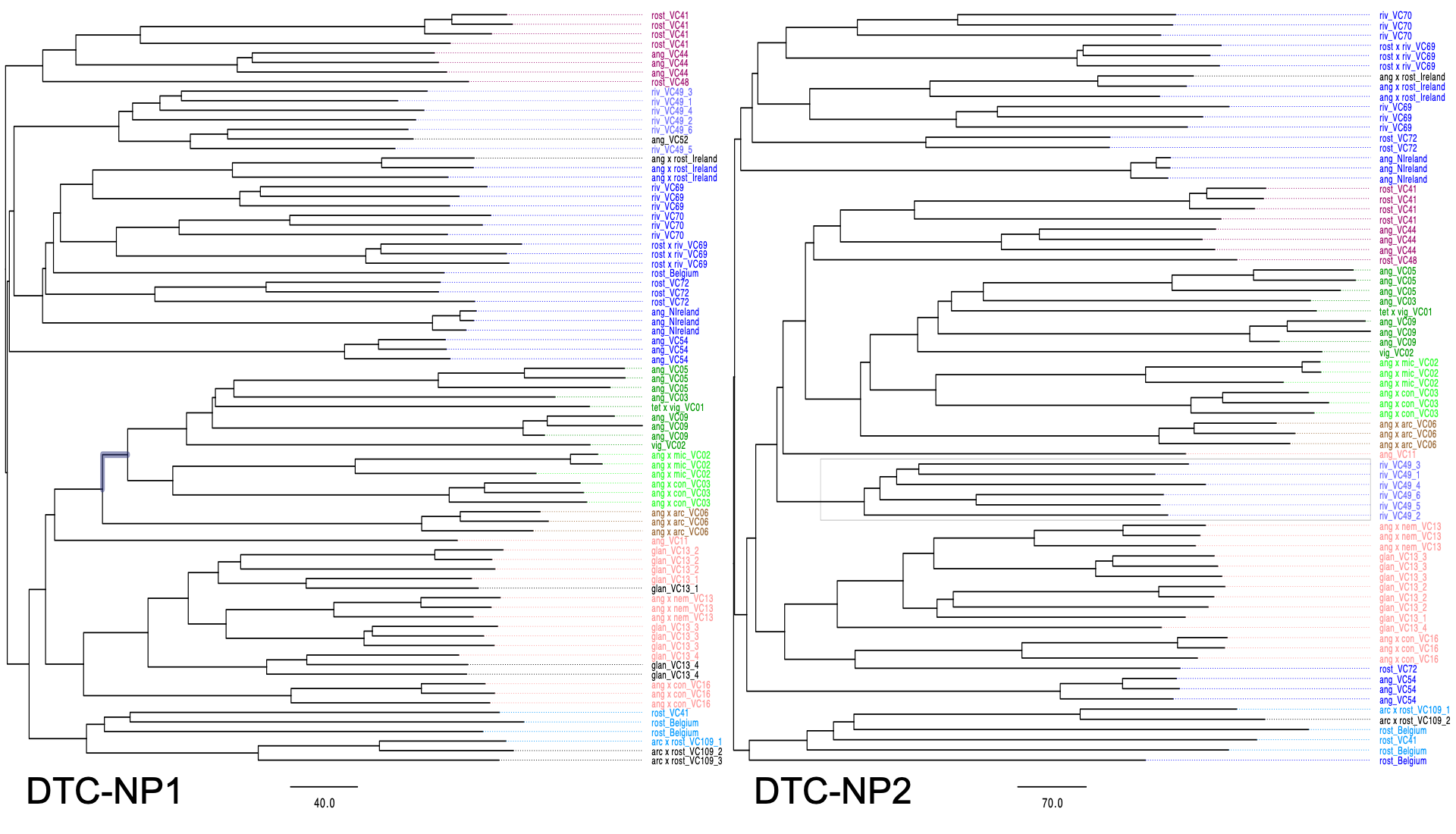
Then, do the DAPC analyses and phylogeny tree using VCF2PopTree, parameters:
- Distance: Genetic distance;
- Missing Data: Use SNVs for each pair of genomes;
- Model: Number of differences;
- Construct Tree: Neighbour-Joining tree (Unrooted);
- Output format: Newick tree
DAPC RESULTS¶

ERROR & SOLUTION INFO¶
1. when in plink, filter SNPs, comes the following error:
Error: More than 4 different alleles at variant 21566.
Solution1: use GATK
##Picard, to produce reference genome for next step:
wget https://github.com/broadinstitute/picard/releases/download/2.22.3/picard.jar
# test it is fine
java -jar /localdisk/home/s1950737/gatk-4.1.6.0/picard.jar -h
## these two are creating reference genome format (.fai):
java -jar picard.jar CreateSequenceDictionary R= ../referenceGenome/tet/scaffolds_disom_concat_dipOrTet.fasta O= tet.dict
samtools faidx ../referenceGenome/tet/scaffolds_disom_concat_dipOrTet.fasta
##index needed:
bgzip -c tet.vcf > tet.vcf.gz
tabix -fp vcf tet.vcf.gz
##select biallelic variants
./gatk SelectVariants -R ../referenceGenome/tet/scaffolds_disom_concat_dipOrTet.fasta -V ../bi-tet/tet2.vcf --restrict-alleles-to BIALLELIC -O ../bi-tet/ouput2.vcf
# didn't work, don't know why
Solution2: use vcftools
vcftools --vcf tet.vcf --min-alleles 2 --max-alleles 2 --recode bi-tet.vcf
vcftools --vcf tet.vcf --min-alleles 1 --max-alleles 2 --recode tet1-2.vcf #try to see the difference